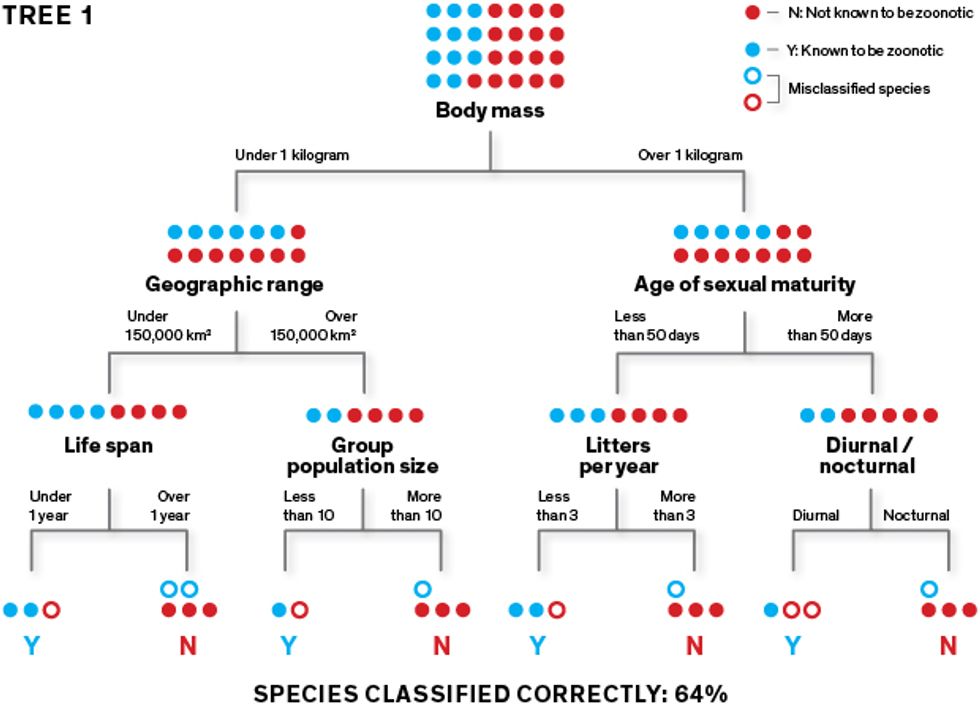
 The algorithm learns how to classify species as “zoonotic” (represented here as “Y”) or “not known to be zoonotic” (represented as “N”) using a training data set. To create an initial classification tree, it repeatedly splits the data set of rodent species into two groups, using a randomly selected feature (such as body size under or over 1 kilogram) for each split. Its goal is to separate the Ys from the Ns at the terminal “leaves” of the tree.
The algorithm learns how to classify species as “zoonotic” (represented here as “Y”) or “not known to be zoonotic” (represented as “N”) using a training data set. To create an initial classification tree, it repeatedly splits the data set of rodent species into two groups, using a randomly selected feature (such as body size under or over 1 kilogram) for each split. Its goal is to separate the Ys from the Ns at the terminal “leaves” of the tree.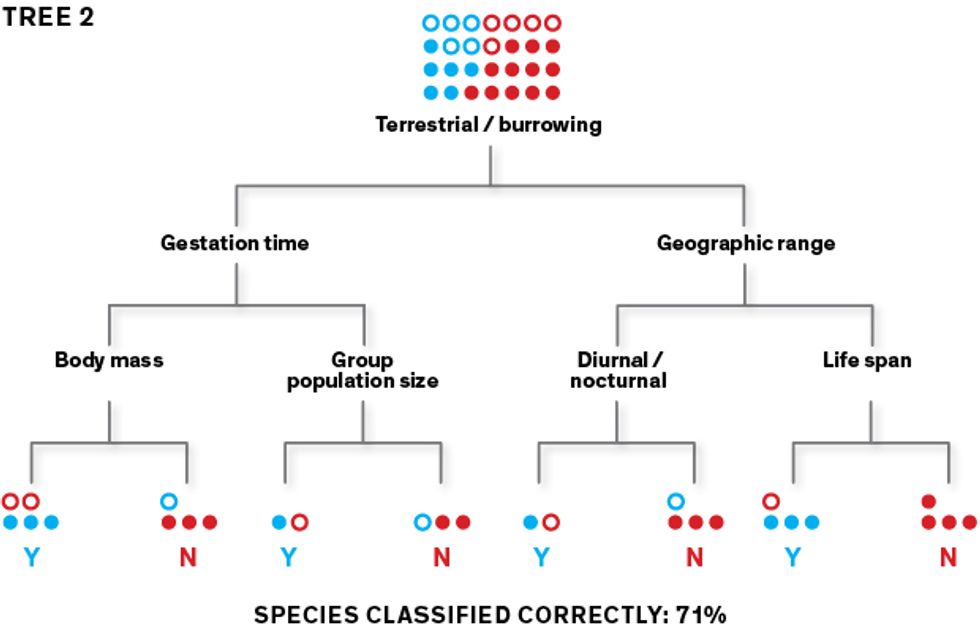 This first tree may produce lots of classification errors, so the algorithm builds a second tree that prioritizes the misclassified species, aiming to sort them correctly. The second tree’s misclassified species are prioritized as the algorithm builds its third tree, and so on.
This first tree may produce lots of classification errors, so the algorithm builds a second tree that prioritizes the misclassified species, aiming to sort them correctly. The second tree’s misclassified species are prioritized as the algorithm builds its third tree, and so on.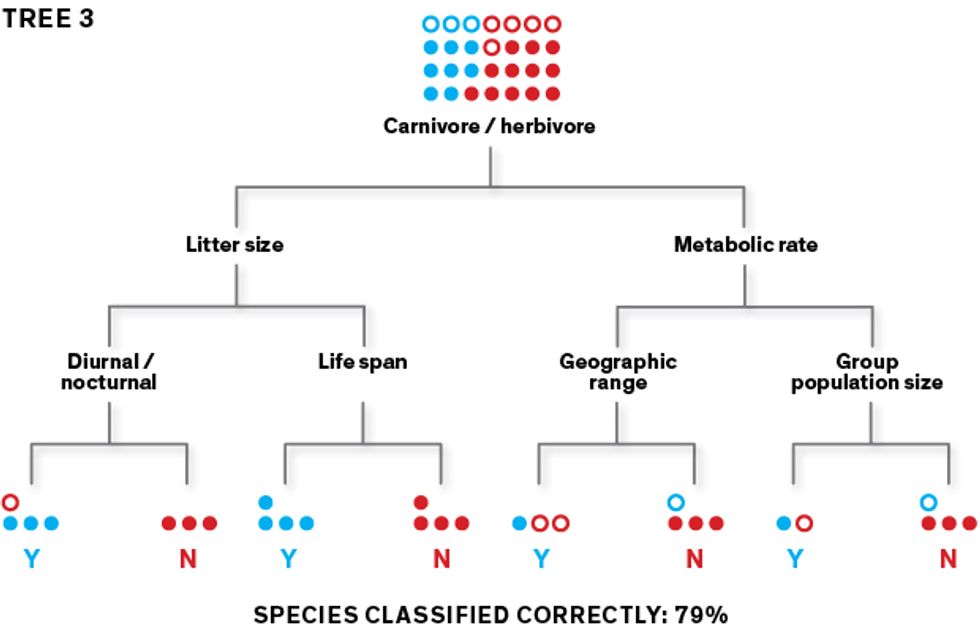 In this iterative fashion, the algorithm generates thousands of trees. When data is filtered through all these trees as an ensemble, the classification accuracy goes way up. Once the model performs well on the training data, we use it to make predictions with the rest of the data set.Illustrations: Erik Vrielink
In this iterative fashion, the algorithm generates thousands of trees. When data is filtered through all these trees as an ensemble, the classification accuracy goes way up. Once the model performs well on the training data, we use it to make predictions with the rest of the data set.Illustrations: Erik VrielinkThe Conversation (0)

Culprit Creature: Humans catch zoonotic diseases through contact with infected animals. This fruit bat is a known carrier of Nipah virus, a potentially deadly disease first identified in Malaysia in 1999.
Photo: Joe McDonald/Getty Images
In April 2014, just after world health officials identified a series of suspicious deaths in Guinea as an outbreak of Ebola, 10 ecologists, 4 veterinarians, and an anthropologist traveled to a Guinean village named Meliandou. Theirs was a detective mission to determine how this outbreak began. How had “patient zero,” a 2-year-old boy named Emile, contracted the Ebola virus?
Because we believe people catch Ebola through contact with infected animals, ecologists have long sought the animal “reservoirs” that harbor the virus and pass it along (often without getting sick themselves). With every new outbreak of a zoonotic disease like Ebola, scientists race to identify the reservoirs so that public health officials can determine the method of transmission and perhaps prevent more “spillover events,” in which the disease flows from animal reservoirs to people. Such is today’s post hoc, reactive model of dealing with outbreaks.
In Meliandou, the Ebola detectives interviewed villagers, studied primate populations in nearby forests, and collected bats in nets. In December 2014, they published a paper hypothesizing that little Emile had contracted Ebola from a colony of insect-eating bats that lived in a hollow tree, near where the local children often played. But the tree had caught fire before the team arrived in the village and the bats were gone, so the investigators couldn’t say for sure.


 The (Probable) Source: Epidemiologists believe the current Ebola outbreak began in the Guinean village of Meliandou (top), when a child playing near a hollow tree (middle) contracted the virus from an Angolan free-tailed bat (bottom).Photos, from top: Kathrin Nowak; Fabian Leendertz; Ivan Kuzmin/Alamy
The (Probable) Source: Epidemiologists believe the current Ebola outbreak began in the Guinean village of Meliandou (top), when a child playing near a hollow tree (middle) contracted the virus from an Angolan free-tailed bat (bottom).Photos, from top: Kathrin Nowak; Fabian Leendertz; Ivan Kuzmin/Alamy
As most previous research on Ebola reservoirs has focused on fruit bats, the team’s findings may prompt scientists to study this insectivorous bat species, and may cause health officials to stay alert in areas where these bats live in close proximity to people. But these are rearguard maneuvers against a brutal opponent: The current Ebola epidemic has killed more than 11,200 people in West Africa to date, and health officials are still fighting to end it. Is there a way to go on the offense against Ebola and other zoonotic diseases? Can we predict outbreaks before they occur?
In my research as a disease ecologist at the Cary Institute of Ecosystem Studies, in Millbrook, N.Y., I use computer modeling and machine learning to predict which wild species are capable of causing future outbreaks. My models create “caricatures” of likely reservoirs, revealing the suite of features that distinguish the unusual species that can harbor microbes dangerous to humans. I then use algorithms to sort through hundreds or thousands of species that have never been checked for zoonotic diseases, and calculate the probability that any given species is a disease reservoir based on its similarity to that caricature. The models give us a list of suspects.
My colleagues and I do this work in the spirit of scientific inquiry, and also with an urgent sense of purpose. Infectious diseases are on the rise around the world, and the U.S. Agency for International Development reckons that about 75 percent of new diseases are zoonotic. If we can predict which species may carry infections capable of jumping to humans, we can monitor the potential hot spots where people interact with these creatures. One day, I hope that biologists will forecast disease outbreaks in the same way meteorologists forecast the weather. With one major difference: A meteorologist can’t stop a storm front, but we may be able to prevent outbreaks.
How a Machine Learns
This very simplified diagram shows how our algorithm creates classification trees, which it can then use to predict which rodent species carry zoonotic diseases.  The algorithm learns how to classify species as “zoonotic” (represented here as “Y”) or “not known to be zoonotic” (represented as “N”) using a training data set. To create an initial classification tree, it repeatedly splits the data set of rodent species into two groups, using a randomly selected feature (such as body size under or over 1 kilogram) for each split. Its goal is to separate the Ys from the Ns at the terminal “leaves” of the tree.
The algorithm learns how to classify species as “zoonotic” (represented here as “Y”) or “not known to be zoonotic” (represented as “N”) using a training data set. To create an initial classification tree, it repeatedly splits the data set of rodent species into two groups, using a randomly selected feature (such as body size under or over 1 kilogram) for each split. Its goal is to separate the Ys from the Ns at the terminal “leaves” of the tree. This first tree may produce lots of classification errors, so the algorithm builds a second tree that prioritizes the misclassified species, aiming to sort them correctly. The second tree’s misclassified species are prioritized as the algorithm builds its third tree, and so on.
This first tree may produce lots of classification errors, so the algorithm builds a second tree that prioritizes the misclassified species, aiming to sort them correctly. The second tree’s misclassified species are prioritized as the algorithm builds its third tree, and so on.- In this iterative fashion, the algorithm generates thousands of trees. When data is filtered through all these trees as an ensemble, the classification accuracy goes way up. Once the model performs well on the training data, we use it to make predictions with the rest of the data set.Illustrations: Erik Vrielink
To understand why the reactive approach to outbreaks has prevailed thus far, just look at Ebola. Imagine you’re a wildlife biologist trying to find this virus’s reservoirs where it first emerged, in the Congo rain forest. You’re standing in front of a forest roughly the size of Alaska that’s home to more than 1,400 species of mammals and birds, as well as countless insect species. If you have the resources, you might try to sample every animal you can catch: many representatives of common species and occasional solitary specimens from rare species.
Even then, you probably won’t succeed in your goal. Only a fraction of a reservoir species’ population will be infected—and given the intermittent nature of Ebola outbreaks, the prevalence of the virus in its animal host populations is thought to be very low. Also, there may be multiple reservoir species—and you’re trying to identify them all in a dynamic environment, where animals migrate with the seasons and relocate due to habitat destruction. Even if you get your hands on an infected animal, the Ebola virus may be hard to spot: The amount of virus in the animal’s body may vary seasonally or according to the animal’s stress levels.
During previous surveys seeking Ebola’s wild reservoirs, biologists have collected more than 30,000 individuals from hundreds of species. While traces of prior infections (that is, antibodies) have been detected in the blood of a number of animals, we have yet to isolate the live virus in the body of a living animal. Biologists won’t give up the search, but clearly an additional approach would be welcome.
In my work, I use machine-learning algorithms that take in vast amounts of unstructured data about wildlife and identify the key traits that are most helpful in predicting a reservoir species. The algorithms I apply are an extension of tools called classification and regression trees, which have been around for decades. The novelty of my work is the application of these techniques to a massive challenge of ecology and global health.
My recent study of rodents, conducted with colleagues at the University of Georgia, gives an example of how this approach works. The order Rodentia includes more than 2,200 species, more than any other group of mammals. Rodents are also prolific with their pathogens: In our conservative estimate, we counted about 200 rodent species that are already known to transmit between 1 and 11 different zoonotic diseases. Among their contributions to human suffering are hantaviruses that can cause fatal pulmonary disease and the bacterium responsible for bubonic plague.
To train our algorithm to find more of these carriers, we fed in data for 80 percent of all rodent species, leaving the remainder to serve later on as a test bed. We gave each species a binary label: a “1” indicating that it’s known to carry a zoonotic disease, or a “0” indicating that its reservoir status is unknown. We also fed in information from sources such as the massive PanTHERIA database of mammals, which collates data from thousands of field studies regarding rodent species’ physiology, behavior, geographic range, social structure, and so forth.
Where to Look for Trouble
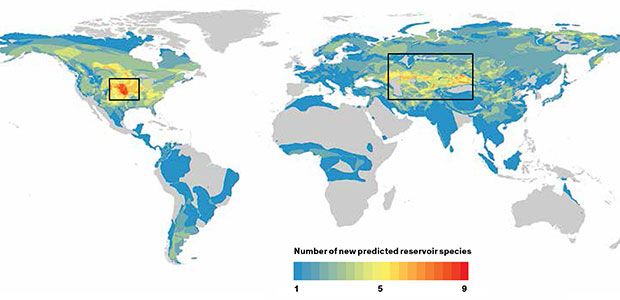
Our computer model identified 58 rodents that had never before been associated with a zoonotic disease as likely “reservoirs” of pathogens that could infect humans. The maps of those rodents’ ranges revealed two potential hot spots where we predict disease outbreaks are likely to occur: the American Midwest and a band across central Asia and the Middle East. Armed with these predictions, field biologists can canvass these areas and study species from our list of suspects, such as the northern grasshopper mouse [below] and the tamarisk jird [bottom].

 Images, from top: Barbara Han; Rick & Nora Bowers/Alamy; Barbara Han; Klaus Rudloff/Moscow Zoo; Barbara Han
Images, from top: Barbara Han; Rick & Nora Bowers/Alamy; Barbara Han; Klaus Rudloff/Moscow Zoo; Barbara Han
The algorithm creates a classification tree by taking the training data and identifying split points: values of particular variables that lead to two classes that are the most different from each other. It does this again and again, creating forking branches until all the data is sorted into a series of bins—the leaves of the classification tree. It can also create a regression tree, which is slightly fancier. Its final leaves don’t simply show binary responses to the split points (such as “one litter per year” versus “more than one litter per year”); instead, its leaves show a continuum of values (such as one, two, three, four litters per year).
In our study, the algorithm generated a tree by randomly selecting a feature to split the group of rodent species into two homogeneous subgroups consisting of “1s” and “0s.” It did this as best it could—inevitably, there were classification errors. It then selected a second feature, then a third, and so on until all the rodents had been separated into leaves of the tree. These features included resting metabolic rate, adult body size, age of sexual maturity, number of offspring per litter, number of litters per year, group population size, and more than 50 other such characteristics.
This method has a major weakness: It’s very sensitive to which feature is selected first. Depending on whether the algorithm selects, say, “group population size” or “metabolic rate” as the first feature, it will produce very different trees. Using any one tree, we won’t have great success in correctly predicting whether a new rodent is a zoonotic suspect or not; its prediction accuracy may be little better than a coin toss. To overcome this fault, we apply an iterative process called “boosting.” Here, the algorithm focuses on the errors it made in any given tree, and prioritizes that data as it creates new trees. This method generates hundreds or thousands of weakly predictive trees that, when employed as an ensemble, produce a powerfully accurate predictive model.
When we tested our rodent-sorting algorithm on the 20 percent of rodents that hadn’t been included in the training data set, it predicted species’ reservoir status with about 90 percent accuracy. And when we pulled back the curtain to see which features the algorithm had used to make its accurate predictions, we saw that it identified zoonotic reservoirs based on a unique trait profile. It had not, as you might expect, picked out species that were closely related to each other. Instead, it found that reservoir species were distinguished by their “fast” life cycles—with rapid growth rates, early sexual maturity, and frequent litters. This finding fits nicely with in-depth studies of individual rodents, which have suggested that reservoir species may have less sensitive immune systems. These animals may tolerate pathogens because they have a “live fast, die young” strategy: Their immune systems aren’t their top priority because they need to stay healthy just long enough to reproduce. The profile also showed that reservoir species tend to have large geographic ranges. These animals may thrive in diverse ecological habitats or may adapt well to the fragmented and heterogeneous landscapes created by humans.
Our study yielded more than scientific insights: It also provided actionable intelligence. As the algorithm sorted through the 2,200 rodent species, it provided a list of new suspects. Some species that had previously been given a “0” for unknown reservoir status fit more neatly in the “1” category of known disease carriers. We didn’t have to wait long for validation. While we were getting our results to press, two of those suspect species were indeed recognized as novel reservoirs for human diseases. One species, a red backed vole (Myodes gapperi) native to Canada and the northern United States, was found to carry the parasite that causes echinococcosis, a nasty ailment in which cysts grow in multiple organs. And researchers identified a vole (Microtus guentheri) native to Asia Minor as a newfound reservoir for leishmaniasis, which causes skin ulcers.
Our list of suspects presents an opportunity for biologists: They can go out into the field and try to “ground-truth” our results. And those field studies will, in turn, inform our work. As surveillance continues and biologists make new discoveries of disease carriers, our databases will grow richer and our model’s predictions will become more accurate. The algorithm will continue to evolve, continue to learn.
We’re now applying our methods to help combat other devastating diseases. We’re currently trying to determine which additional bat species may be reservoirs for the filoviruses that cause hemorrhagic fevers such as Ebola and Marburg virus disease. We hope our results will help explain how certain bats can live with an infection that’s so deadly to great apes, including humans.
Already our model has identified a cluster of bat species that belong on a watch list. To our surprise, some of the species that seem capable of carrying Ebola-like viruses live outside of Africa, in countries where human outbreaks of hemorrhagic fevers have never been officially reported. The results raise a question for biologists: If outbreaks truly haven’t occurred in these places, why not? They also raise a question for public health officials: Should they be worried?
Machine-learning methods have a few key advantages for ecology, a discipline that seeks to understand the complex and ever-shifting interplay between the billions of living beings jockeying for position on Earth.
For instance, our algorithm can deal with our incomplete data sets. Biologists simply can’t learn everything about the 1.6 million species we’ve cataloged thus far, let alone the many millions we haven’t. But the algorithm considers the presence or absence of any particular piece of data as just another variable that can be used as a split point in its classification trees.
Moreover, our approach counteracts the sampling bias that can skew the study of infectious diseases: Extensive wildlife surveys in wealthy regions like the United States and Europe have resulted in higher-quality data regarding American and European species. Biologists can also fall afoul of vigilance bias when they study individual host species: The more they look for something, the more likely they will find it. Thus, if they find that the Norwegian rat carries disease X, they are likely to also sample it for Y and Z, with the result that a few species may appear downright plague ridden, while others have yet to be checked for any pathogen.
Some of the species that seem capable of carrying Ebola-like viruses live outside of Africa, in countries where outbreaks have never been reported
As our method focuses on intrinsic properties of species, we minimize the effects of such biases. For example, if the algorithm zeros in on rodent species with small bodies, it will draw species from all over the world (because small-bodied rodents are just as likely to live in poor countries as rich ones). By using species’ intrinsic biology to predict reservoir status, we avoid falling into the vigilance bias trap: making predictions that center on places that can afford surveillance to begin with. On the other hand, there isn’t much to be done about serious data deficiency. If we have no data on a species, there’s simply no possibility of predicting its reservoir probability. Our work shows that cutting basic science funding has significant ripple effects: It really is worth knowing the life history of that obscure mouse in Papua New Guinea.
Machine learning also deals well with complexity. Ecological analyses can easily include dozens of variables, but it’s often not clear how those variables interact. For instance, although there’s good evidence that an animal’s body size and metabolic rate scale according to a particular mathematical relationship, it’s less clear how the size of a newborn relates to metabolic rate. The more variables there are, the harder it is to understand their complex and hidden interactions.
But our algorithm doesn’t require us to set any rules for these interactions. Instead, our method allows the data to speak for itself. If a particular combination of variables leads to great predictive accuracy, the model identifies those variables and presents them to the researcher for further interpretation. The algorithm doesn’t care how the variables are interacting; its only goal is to maximize predictive performance. Then we human scientists can step up. Our job is to look at the variables that are most important for prediction and figure out what they reveal about the biology of zoonotic disease reservoirs.
To make progress toward the ambitious goal of forecasting and even preempting zoonotic disease outbreaks, it’s not enough to learn which disease arises from contact with which reservoir. Biologists need to understand: Why is that particular species special? Our approach gives us a clue about the “why”—the biological mechanisms that allow some animals to be carriers and transmitters of deadly infections.
Of course, human beings play a role in the emergence of disease, often by coming directly into contact with wild animals or by bringing domestic animals into contact with them. For example, Nipah virus emerged in Malaysia from human contact with infected pigs, which had picked up the virus from fruit bats. Those bats had begun foraging in orchards and swine farms because people had chopped down their forest habitats.
Urbanization, deforestation, and hunting will continue to bring humans into contact with wild species potentially bearing wild new diseases. We’re all part of the same system of life, and diseases emerge from this complex system. We’re only beginning to understand these ecological dynamics. Predicting reservoir species is quite a challenge, but I see it as part of an even greater challenge: figuring out how to live harmoniously with the wild creatures with which we share this planet
This article originally appeared in print as “Building a Better Disease Detective.”
About the Author
Barbara Han is a research scientist at the Cary Institute of Ecosystem Studies, in Millbrook, N.Y. While she has done biological fieldwork in the past, these days she interacts with animals through her data sets. She’s somewhat surprised to have ended up as a computer programmer, but there are aspects of fieldwork that she doesn’t miss. “I like working with animals, but I don’t like being bitten by animals,” Han says.


