
In June 2000, a press conference was held in the White House to announce an extraordinary feat: the completion of a draft of the human genome.
For the first time, researchers had read all 3 billion of the chemical “letters” that make up a human DNA molecule, which would allow geneticists to investigate how that chemical sequence codes for a human being. In his remarks, President Bill Clinton recalled the moment nearly 50 years prior when Francis Crick and James Watson first discovered the double-helix structure of DNA. “How far we have come since that day,” Clinton said.
But the president’s comment applies equally well to what has happened in the ensuing years. In little more than a decade, the cost of sequencing one human genome has dropped from hundreds of millions of dollars to just a few thousand dollars. Instead of taking years to sequence a single human genome, it now takes about 10 days to sequence a half dozen at a time using a high-capacity sequencing machine. Scientists have built rich catalogs of genomes from people around the world and have studied the genomes of individuals suffering from diseases; they are also making inventories of the genomes of microbes, plants, and animals. Sequencing is no longer something only wealthy companies and international consortia can afford to do. Now, thousands of benchtop sequencers sit in laboratories and hospitals across the globe.
DNA sequencing is on the path to becoming an everyday tool in life-science research and medicine. Institutions such as the Mayo Clinic and the New York Genome Center are beginning to sequence patients’ genomes in order to customize care according to their genetics. For example, sequencing can be used in the diagnosis and treatment of cancer, because the pattern of genetic abnormalities in a tumor can suggest a particular course of action, such as a certain chemotherapy drug and the appropriate dose. Many doctors hope that this kind of personalized medicine will lead to substantially improved outcomes and lower health-care costs.

But while much of the attention is focused on sequencing, that’s just the first step. A DNA sequencer doesn’t produce a complete genome that researchers can read like a book, nor does it highlight the most important stretches of the vast sequence. Instead, it generates something like an enormous stack of shredded newspapers, without any organization of the fragments. The stack is far too large to deal with manually, so the problem of sifting through all the fragments is delegated to computer programs. A sequencer, like a computer, is useless without software.
But there’s the catch. As sequencing machines improve and appear in more laboratories, the total computing burden is growing. It’s a problem that threatens to hold back this revolutionary technology. Computing, not sequencing, is now the slower and more costly aspect of genomics research. Consider this: Between 2008 and 2013, the performance of a single DNA sequencer increased about three- to fivefold per year. Using Moore’s Law as a benchmark, we might estimate that computer processors basically doubled in speed every two years over that same period. Sequencers are improving at a faster rate than computers are. Something must be done now, or else we’ll need to put vital research on hold while the necessary computational techniques catch up—or are invented.
How can we help scientists and doctors cope with the onslaught of data? This is a hot question among researchers in computational genomics, and there is no definitive answer yet. What is clear is that it will involve both better algorithms and a renewed focus on such “big data” approaches as parallelization, distributed data storage, fault tolerance, and economies of scale. In our own research, we’ve adapted tools and techniques used in text compression to create algorithms that can better package reams of genomic data. And to search through that information, we’ve borrowed a cloud computing model from companies that know their way around big data—companies like Google, Amazon.com, and Facebook.
Think of a DNA molecule as a string of beads. Each bead is one of four different nucleotides: adenine, thymine, cytosine, or guanine, which biologists refer to by the letters A, T, C, and G. Strings of these nucleotides encode the building instructions and control switches for proteins and other molecules that do the work of maintaining life. A specific string of nucleotides that encodes the instructions for a single protein is called a gene. Your body has about 22 000 genes that collectively determine your genetic makeup—including your eye color, body structure, susceptibility to diseases, and even some aspects of your personality.
Thus, many of an organism’s traits, abilities, and vulnerabilities hinge on the exact sequence of letters that make up the organism’s DNA molecule. For instance, if we know your unique DNA sequence, we can look up information about what diseases you’re predisposed to, or how you will respond to certain medicines.

The Human Genome Project’s goal was to sequence the 3 billion letters that make up the genome of a human being. Because humans are more than 99 percent genetically identical, this first genome has been used as a “reference” to guide future analyses. A larger, ongoing project is the 1000 Genomes Project, aimed at compiling a more comprehensive picture of how genomes vary among individuals and ethnic groups. For the U.S. National Institutes of Health’s Cancer Genome Atlas, researchers are sequencing samples from more than 20 different types of tumors to study how the mutated genomes present in cancer cells differ from normal genomes, and how they vary among different types of cancer.
Ideally, a DNA sequencer would simply take a biological sample and churn out, in order, the complete nucleotide sequence of the DNA molecule contained therein. At the moment, though, no sequencing technology is capable of this. Instead, modern sequencers produce a vast number of short strings of letters from the DNA. Each string is called a sequencing read, or “read” for short. A modern sequencer produces reads that are a few hundred or perhaps a few thousand nucleotides long.
The aggregate of the millions of reads generated by the sequencer covers the person’s entire genome many times over. For example, the HiSeq 2000 machine, made by the San Diego–based biotech company Illumina, is one of the most powerful sequencers available. It can sequence roughly 600 billion nucleotides in about a week—in the form of 6 billion reads of 100 nucleotides each. For comparison, an entire human genome contains 3 billion nucleotides. And the human genome isn’t a particularly long one—a pine tree genome has 24 billion nucleotides.
Thus our first daunting task upon receiving the reads is to stitch them together into longer, more interpretable units, such as genes. For a organism that has never been fully sequenced before, like the pine tree, it’s a massive challenge to assemble the genome from scratch, or de novo.
How can we assemble a genome for the first time if we have no knowledge of what the finished product should look like? Imagine taking 100 copies of the Charles Dickens novel A Tale of Two Cities and dropping them all into a paper shredder, yielding a huge number of snippets the size of fortune-cookie slips. The first step to reassembling the novel would be to find snippets that overlap: “It was the best” and “the best of times,” for example. A de novo assembly algorithm for DNA data does something analogous. It finds reads whose sequences “overlap” and records those overlaps in a huge diagram called an assembly graph. For a large genome, this graph can occupy many terabytes of RAM, and completing the genome sequence can require weeks or months of computation on a world-class supercomputer.
De Novo Sequencing
To sequence a species’ genome for the first time, the double-stranded DNA molecule is first split down the middle [1]. This creates a single-stranded template. The template is copied many times [2]. All the templates are then diced up at random, to create many small fragments of different lengths [3]. A sequencing machine determines the order of nucleotides on each of those short fragments [4]. Then a software program looks for the places where the sequences of letters overlap and constructs an “assembly graph” [5]. Finally, the program constructs one continuous sequence of letters: the genome [6].
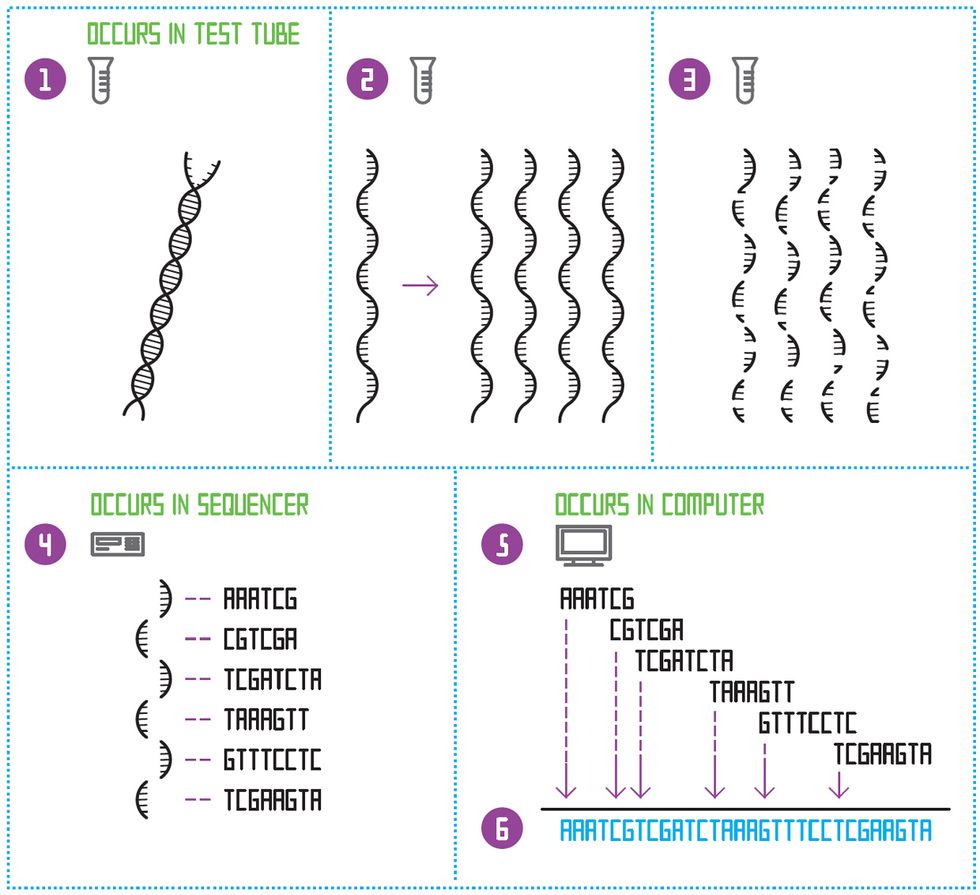 Illustration: L-Dopa
Illustration: L-Dopa
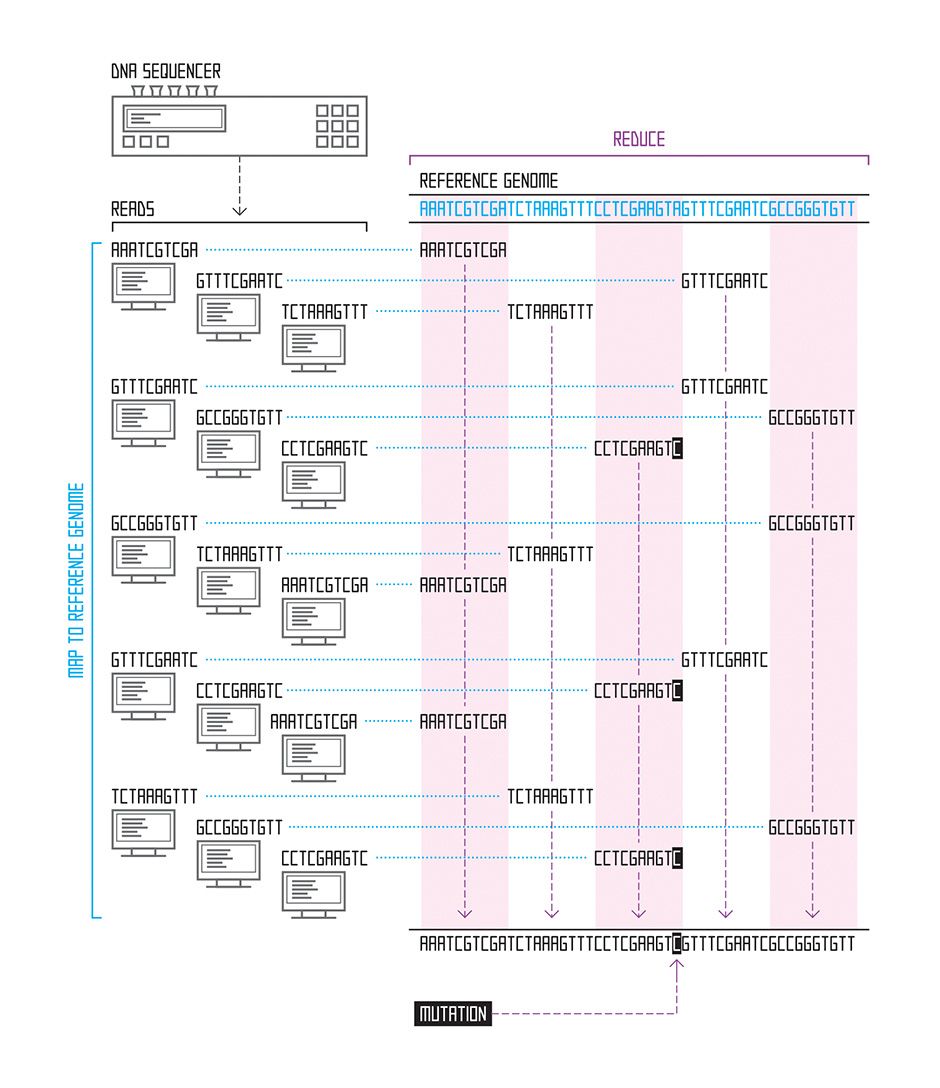
We have an easier job when we’re studying a species whose genome has already been assembled. If we’re examining mutations in human cancer genomes, for example, we can download the previously assembled human genome from the National Institutes of Health website and use it as a reference. For each read, we find the point where that string of letters best matches the genome, using an approximate matching algorithm; the process is similar to how your spell-check program finds the correct spelling based on your misspelled word. The place where the read sequence most closely matches the reference sequence is our best guess as to where it belongs. Thanks to the Human Genome Project and similar projects for other species (mouse, fruit fly, chicken, cow, and thousands of microbial species, for example), many assembled genomes are available for use as references for this task, which is called read alignment.
In general, these reference genomes are far too long for brute force scanning algorithms—those that simply start at the beginning of the sequence and work their way through the entire genome, looking for the part that best matches the read in question. Instead, researchers have lately focused on building an effective genome index, which allows them to rapidly home in on only those portions of the reference genome that contain good matches. Just like an index at the back of a book, a genome index is a list of all the places in the genome where a certain string of letters appears—for example, the roughly 697 000 occurrences of the sequence “GATTACA” in the human genome.
One powerful recent invention is a genome index based on the Burrows-Wheeler transform—an algorithm originally developed for text compression. This efficient index allows us to align many thousands of 100-nucleotide reads per second. The algorithm works by carefully changing the order of a sequence of letters into one that’s more compressible—and doing so in a way that’s reversible. So, for example, let’s say you have 21 As in your jumbled string of As, Ts, Gs, and Cs. That part of the string could then be compressed into A21, thus using 3 characters instead of 21—a sevenfold savings. By compiling a genome index of sequences reordered in this way, the search algorithm can scroll through the entire genome much more quickly, looking for a read’s best match.
Once we have the best algorithms and data structures, we arrive at the next massive challenge: scaling up, and getting many computers to divvy up the work of parsing a genome.
The roughly 2000 sequencing instruments in labs and hospitals around the world can collectively sequence 15 quadrillion nucleotides per year, which equals about 15 petabytes of compressed genetic data. A petabyte is 250 bytes, or in round numbers, 1000 terabytes. To put this into perspective, if you were to write this data onto standard DVDs, the resulting stack would be more than 2 miles tall. And with sequencing capacity increasing at a rate of around three- to fivefold per year, next year the stack would be around 6 to 10 miles tall. At this rate, within the next five years the stack of DVDs could reach higher than the orbit of the International Space Station.
Clearly, we’re dealing with a data deluge in genomics. This data is vital for the advancement of biology and medicine, but storing, analyzing, and sharing such vast quantities is an immense challenge. Still, it’s not an unprecedented one: Other fields, notably high-energy physics and astronomy, have already encountered this problem. For example, the four main detectors at the Large Hadron Collider produced around 13 petabytes of data in 2010, and when the Large Synoptic Survey Telescope comes on line in 2016, it’s anticipated to produce around 10 petabytes per year.
The crucial difference is that these physics and astronomy data deluges pour forth from just a few major instruments. The DNA data deluge comes from thousands—and soon, tens of thousands—of sources. After all, almost any life-science laboratory can now afford to own and operate a sequencer. Major centers like the Broad Institute, in Cambridge, Mass., or BGI, in Shenzhen, China, have more than 100 high-capacity instruments on site, but smaller institutions like the Malaysia Genome Institute or the International Livestock Research Institute, in Kenya, also have their own instruments. In all these facilities, researchers are struggling to analyze the sequencing data for a wide variety of applications, such as investigations into human health and disease, plant and animal breeding, and monitoring microbial ecology and pathogen outbreaks.
The only hope for these overwhelmed researchers lies in advanced computing technologies. Genomics researchers are investigating a range of options, including very powerful but conventional servers, specialized hardware, and cloud computing. Each has strengths and weaknesses depending on the specific application and analysis. But for many, cloud computing is increasingly the best option, because it allows the close integration of powerful computational resources with extremely high-volume data storage.
One promising solution comes from Google, a company with plenty of experience searching vast troves of data. Google doesn’t regularly release information on how much data it processes, but in May 2010 it reported searching 946 petabytes per month. Today, three years later, it’s safe to assume that figure is at least an order of magnitude larger.
To mine the Internet, Google developed a parallel computing framework called MapReduce. Outside of Google, an open-source alternative to MapReduce called Apache Hadoop is emerging as a standard platform for analyzing huge data sets in genomics and other fields. Hadoop’s two main advantages are its programming model, which harnesses the power of many computers in tandem, and its smart integration of storage and computational power.
While Hadoop and MapReduce are simple by design, their ability to coordinate the activity of many computers makes them powerful. Essentially, they divide a large computational task into small pieces that are distributed to many computers across the network. Those computers perform their jobs (the “map” step), and then communicate with each other to aggregate the results (the “reduce” step). This process can be repeated many times over, and the repetition of computation and aggregation steps quickly produces results. This framework is much more powerful than basic “queue system” software packages like the widely used HTCondor and Grid Engine. These systems also divide up large tasks among many computers but make no provision for the computers to exchange information.
Hadoop has another advantage: It uses the computer cluster’s computational nodes for data storage as well. This means that Hadoop can often execute programs on the nodes themselves, thus moving the code to the data rather than having to access data in a comparatively slow file server. This structure also brings a reliability bonus, even on off-the-shelf servers and disks. Google created MapReduce to run in data centers packed with cheap commodity computers, some of which were expected to fail every day, so fault tolerance was built into the system. When a data set is loaded into the program, it’s split up into manageable chunks, and each chunk is replicated and sent to several computer nodes. If one fails, the others go on. This model also works well in a flexible setting such as the Amazon Elastic Compute Cloud, where nodes can be provisioned for an application as needed, on the fly, and leased on a per-hour basis.
We’re still a long way from having anything as powerful as a Web search engine for sequencing data, but our research groups are trying to exploit what we already know about cloud computing and text indexing to make vast sequencing data archives more usable. Right now, agencies like the National Institutes of Health maintain public archives containing petabytes of genetic data. But without easy search methods, such databases are significantly underused, and all that valuable data is essentially dead. We need to develop tools that make each archive a useful living entity the way that Google makes the Web a useful living entity. If we can make these archives more searchable, we will empower researchers to pose scientific questions over much larger collections of data, enabling greater insights.
This year, genomics researchers may reach a remarkable milestone: the US $1000 genome. Experts have long said that when the cost of sequencing a human genome falls to that level, the technology can be used routinely in biological research and medical care. The high-capacity Illumina systems are nearing this price point, as is the Ion Proton machine from San Diego–based Life Technologies.
Such sequencing capacity is already enabling projects that can reinvent major sectors of technology, science, and medicine. For example, the U.S. Department of Energy recently launched KBase, a knowledge base for biofuel research that integrates hundreds of terabytes of genomic and other biological data inside its own compute cloud. KBase will use state-of-the-art machine learning and data-mining techniques to build predictive models of how genome variations influence the growth of plants and microbes in different environments. Researchers can then select which plants and microbes should be bred or genetically engineered to become more robust, or to produce more usable oils.
This scenario is just a hint of what is to come if we can figure out how to channel the data deluge in genomics. As sequencing machines spew out floods of As, Ts, Cs, and Gs, software and hardware will determine how much we all benefit.
About the Authors
Michael C. Schatz is an assistant professor of quantitative biology at Cold Spring Harbor Laboratory, in New York state. Schatz knows firsthand how much genetics research depends on computer science. His first postcollege job was working as a software engineer in the trenches of a major genomics research institute, where he wrote programs for analyzing all the genetic data generated by high-tech sequencing machines.
Ben Langmead began collaborating with Schatz when the two were Ph.D. students in bioinformatics and computational biology. Now an assistant professor of computer science at Johns Hopkins University, Langmead relishes the contributions his programs can make to medical and scientific research. “When I’m helping life scientists design an experiment so the data they get can be analyzed, it answers the ‘What’s the point?’ question,” he says.