The Conversation (1)

John Bavaro/Science Source
Since the beginning of 2020, we've heard an awful lot about RNA. First, an RNA coronavirus created a global pandemic and brought the world to a halt. Scientists were quick to sequence the novel coronavirus's genetic code, revealing it to be a single strand of RNA that is folded and twisted inside the virus's lipid envelope. Then, RNA vaccines set the world back in motion. The first two COVID-19 vaccines to be widely approved for emergency use, those from Pfizer-BioNTech and Moderna, contained snippets of coronavirus RNA that taught people's bodies how to mount a defense against the virus.
But there's much more we need to know about RNA. RNA is most typically single-stranded, which means it is inherently less stable than DNA, the double-stranded molecule that encodes the human genome, and it's more prone to mutations. We've seen how the coronavirus mutates and gives rise to dangerous new variants. We must therefore be ready with new vaccines and booster shots that are precisely tailored to the new threats. And we need RNA vaccines that are more stable and robust and don't require extremely low temperatures for transport and storage.
That's why it's never been more important to understand RNA's intricate structure and to master the ability to design sequences of RNA that serve our purposes. Traditionally, scientists have used techniques from computational biology to tease apart RNA's structure. But that's not the only way, or even the best way, to do it. Work at my group at Baidu Research USA and Oregon State University has shown that applying algorithms originally developed for natural language processing (NLP)—which helps computers parse human language—can vastly speed up predictions of RNA folding and the design of RNA sequences for vaccines.
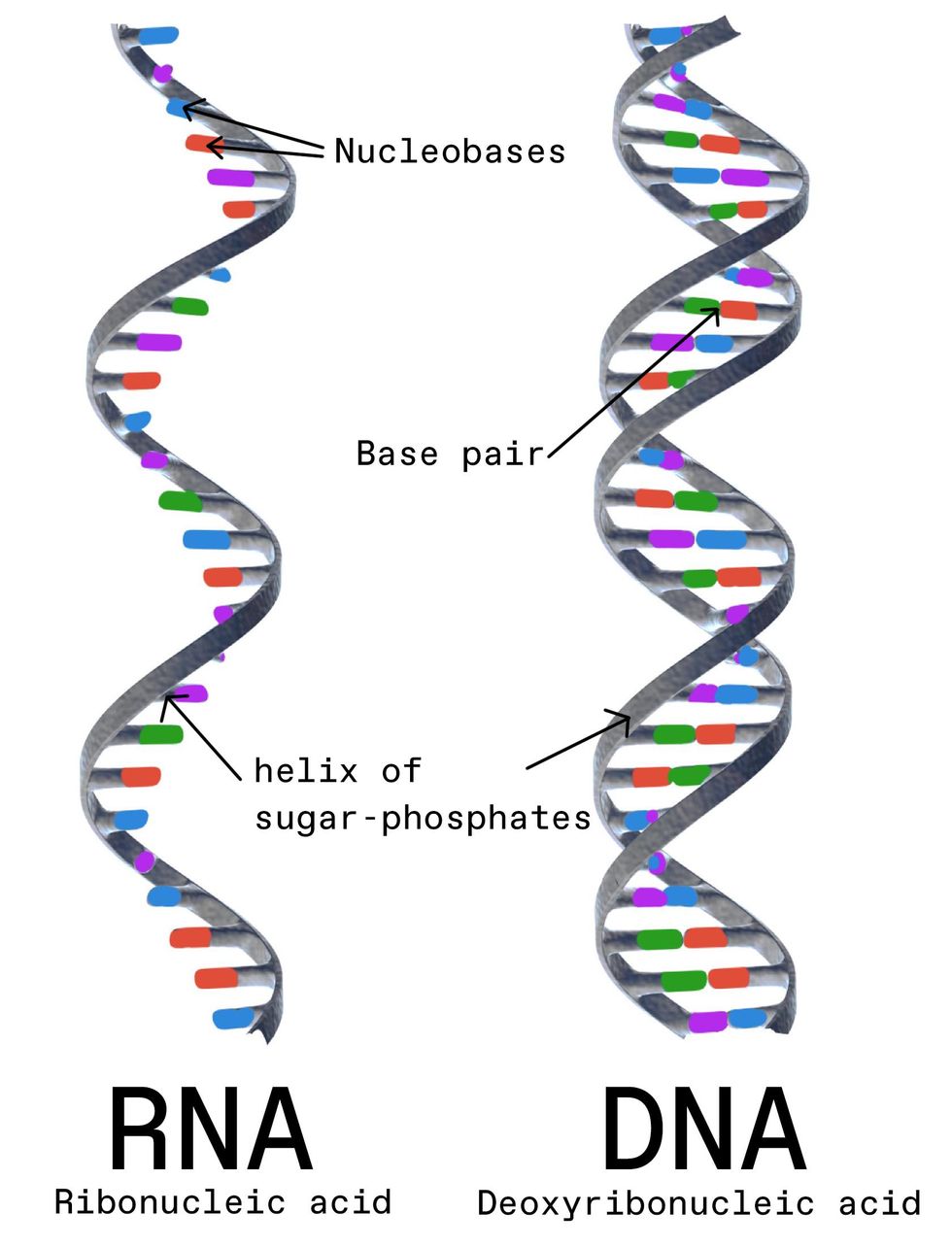 RNA is a single-stranded molecule composed of nucleobases. It's more prone to mutations than DNA, in which nucleobases pair up to create a double-stranded molecule. Gunilla Elam/Science Source
RNA is a single-stranded molecule composed of nucleobases. It's more prone to mutations than DNA, in which nucleobases pair up to create a double-stranded molecule. Gunilla Elam/Science Source
The fields of NLP (also known as computational linguistics) and computational biology may seem very different, but mathematically speaking, they're quite similar. An English-language sentence is made of words that form a sequence. On top of that sequence, there's a structure, a syntactic tree that includes noun phrases and verb phrases. Those two components—the sequence and the structure—together yield meaning. Similarly, a strand of RNA is made up of a sequence of nucleotides, and on top of that sequence, there's the secondary structure of how the strand is folded up.
In English, you can have two words that are far apart in the sentence, but closely linked in terms of grammar. Take the sentence "What do you want to serve the chicken with?" The words "what" and "with" are far apart, but "what" is the object of the preposition "with." Similarly, in RNA you can have two nucleotides that are far apart on the sequence, but close to each other in the folded structure.
My lab has exploited this similarity to adapt NLP tools to the pressing needs of our time. And by joining forces with researchers in computational biology and drug design, we've been able to identify promising new candidates for RNA COVID-19 vaccines in an astonishingly short period of time.
My lab's recent advances in RNA folding build directly on a natural-language processing technique I pioneered called incremental parsing. Humans use incremental parsing constantly: As you're reading this sentence, you're building its meaning in your mind without waiting until you reach the period. But for many years, computers doing a similar comprehension task didn't use incremental parsing. The problem was that language is full of ambiguities that can confound NLP programs. So-called garden-path sentences such as "The old man the boat" and "The horse raced past the barn fell" show how confusing things can get.
Incorrect

Erik Vrielink
Correct

So-called "garden-path sentences" lead the reader in the wrong direction, and also confuse natural-language processing algorithms. In the correct parsing of this sentence [right], the word "man" is a verb.
As a sentence gets longer, the number of possible meanings multiplies. That's why classical NLP parsing algorithms weren't linear—that is, the length of time they took to understand a sentence didn't scale in a linear fashion with the length of a sentence. Instead, comprehension time scaled cubically with sentence length, so that if you doubled the length of a sentence, it took 8 times longer to parse it. Fortunately, most sentences aren't very long. A sentence in English speech is rarely more than 20 words, and even those in The Wall Street Journal are typically under 40 words long. So while cubic time made things slow, it didn't create intractable problems for classical NLP parsing algorithms. When I developed incremental parsing in 2010, it was recognized as an advance but not a game changer.
When it comes to RNA, however, length is a huge problem. RNA sequences can be staggeringly long: The coronavirus genome contains some 30,000 nucleotides, making it the longest RNA virus we know. Classical techniques to predict RNA folding, being almost identical to classical NLP parsing algorithms, were also ruled by cubic time, which made large-scale predictions impractical.
The fields of natural language processing and computational biology may seem very different, but mathematically speaking, they're quite similar.
In late 2015, a chance conversation with a colleague in Oregon State's biophysics department made me notice the similarities between dilemmas in NLP and RNA. That's when I realized that incremental parsing could have a much larger impact in computational biology than it had in my original field.
The old-fashioned NLP technique for parsing sentences was "bottom up," meaning that a parsing program would look first at pairs of consecutive words within the sentence, then sets of three consecutive words, then four, and so on until it was considering the entire sentence.
My incremental parser dealt with language's ambiguities by scanning from left to right through a sentence, constructing many possible meanings for that sentence as it went. When it reached the end of the sentence, it chose the meaning that it deemed most likely. For example, for the sentence "John and Mary wrote two papers each," most of its initial hypotheses about the meaning of the sentence would consider John and Mary as a collective noun phrase; only when it reached the last word—the distributive pronoun "each"—would an alternative hypothesis gain prominence, in which John and Mary are considered separately. With this technique, the time required for parsing scaled in a linear fashion to the length of the sentence.
One significant difference between linguistics and biology is the amount of meaning contained in each piece of the sequence. Each English word carries a lot of meaning; even a simple word like "the" signals the arrival of a noun phrase. And there are many different words in total. RNA strings, by contrast, contain only the four nucleotides adenine, cytosine, guanine, and uracil, with each nucleotide on its own carrying little information. That's why predicting the structure of RNA from its sequence has long been a huge challenge in bioinformatics.
My collaborators and I used the principle of incremental parsing to develop the LinearFold algorithm for predicting RNA structure, which considers many possible structures in parallel as it scans the RNA sequence of nucleotides. Because there are many more possible secondary structures in a long RNA sequence than there are in an English-language sentence, the algorithm considers billions of alternatives for each sequence.
 RNA molecules fold into a complex structure. RNA structure can be depicted graphically [top left] to show nucleotides that pair up and those in “loops" that are unpaired. The same sequence is depicted with lines showing paired nucleotides [top right]; read counter-clockwise, the initial “GCGG" corresponds to the “GCGG" at the top left of the graphical representation. The LinearFold algorithm [bottom] scans the sequence from left to right and tags each nucleotide as unpaired, to be paired with a future nucleotide, or paired with a previous nucleotide.Huang Liang
RNA molecules fold into a complex structure. RNA structure can be depicted graphically [top left] to show nucleotides that pair up and those in “loops" that are unpaired. The same sequence is depicted with lines showing paired nucleotides [top right]; read counter-clockwise, the initial “GCGG" corresponds to the “GCGG" at the top left of the graphical representation. The LinearFold algorithm [bottom] scans the sequence from left to right and tags each nucleotide as unpaired, to be paired with a future nucleotide, or paired with a previous nucleotide.Huang Liang
In 2019, before the start of the pandemic, we published a paper about LinearFold, which we were proud to report was (and still is) the world's fastest algorithm for predicting RNA's secondary structure. In January 2020, when COVID-19 was taking hold in China, we began to think hard about how to apply our work to the world's most pressing problem. The following month, we tested the algorithm with an analysis of SARS-CoV-2, the virus that causes COVID-19. While standard computational biology methods took 55 minutes to identify the structure, LinearFold did the job in only 27 seconds. We built a web server to make the algorithm freely accessible to scientists studying the virus or working on pandemic response. But we weren't done yet.
Understanding how the SARS-CoV-2 virus folds up is useful for basic scientific research. But as the pandemic began to ravage the world, we felt called to help more directly with the response. I reached out to my friend Rhiju Das, an associate professor of biochemistry at Stanford University School of Medicine and a long-time user of LinearFold. Das specializes in computer modeling and design of RNA molecules, and he had created the popular Eterna game, which crowdsources intractable RNA design problems to 250,000 online players. In Eterna challenges, players are presented with a desired RNA structure and asked to find sequences that fold into that shape. Players have worked on RNA sequences for a diagnostic device for tuberculosis and for CRISPR gene editing.
Das was already using LinearFold to speed up the processing of players' designs. In response to the pandemic, he decided to launch a new Eterna challenge called OpenVaccine, asking players to design potential RNA vaccines that would be more stable than existing RNA vaccines. (The RNAs in these vaccines is a particular type called messenger RNA or mRNA for short, hence these vaccines are more formally called mRNA vaccines, but I'll just call them RNA vaccines for simplicity's sake).
Today's RNA vaccines require extremely cold temperatures during transport and storage to remain viable, which has led to vaccines being discarded after power outages and limited their use in hot places where cold-chain infrastructure is lacking, such as India, Brazil, and Africa. If Eterna's players could design a more robust and stable vaccine, it could be a boon for many parts of the world. The OpenVaccine challenge again used LinearFold to speed up processing, but I wondered if it would be possible to develop an algorithm that would do more—that would design the RNA structures directly. Das thought it was a long shot, but I got to work on an algorithm that I called LinearDesign.
 The SARS-CoV-2 virus has spike proteins that hook onto human cells to gain entrance. RNA vaccines for the coronavirus typically contain snippets of RNA that code for just the production of the spike protein, so the immune system can learn to recognize it.N. Hanacek/NIST
The SARS-CoV-2 virus has spike proteins that hook onto human cells to gain entrance. RNA vaccines for the coronavirus typically contain snippets of RNA that code for just the production of the spike protein, so the immune system can learn to recognize it.N. Hanacek/NIST
RNA vaccines for COVID-19 work because they contain a snippet of coronavirus RNA—typically, a snippet that codes for production of the spike protein, the part of the virus that hooks onto human cells to gain entry. Because these vaccines only code for that one protein and not the entire virus, they pose no risk of infection. But when human cells begin to produce that spike protein, it triggers an immune reaction, which ensures that the immune system will be ready if exposed to the real virus. So the challenge for Eterna players was to design more stable RNA snippets that would still code for the spike protein.
Earlier, I said RNA folds up on itself, pairing some complementary nucleotides to produce double-stranded regions, and the unpaired regions remain single-stranded. Those double-strand parts are inherently more stable than single-strand regions, and are less likely to break down inside cells.
Moderna, one of the makers of today's leading RNA vaccines, published a paper in 2019 stating that a more stable secondary structure led to longer-lasting RNA strands, and thus to greater production of proteins—and potentially a more potent vaccine. But relatively little work has been done since then on designing more stable RNA sequences for vaccines. As the pandemic took hold, it seemed clear that optimizing RNA vaccines for greater stability could have huge benefits, so that's what the players of OpenVaccine set out to accomplish.
If Eterna's players could design a more robust and stable vaccine, it could be a boon for many parts of the world.
It was a massive challenge because of some basic biological facts. The coronavirus spike protein is composed of more than 1,000 amino acids, and most amino acids can be encoded by multiple codons. The amino acid glycine is encoded by four different codons (GGU, GGC, GGA, and GGG), the amino acid leucine is encoded by six different codons, and so forth. Because of that redundancy, there are a dizzying number of possible RNA sequences that encode the spike protein—about 2.4 x 10632! In other words, a COVID-19 vaccine has roughly 2.4 x 10632 candidates. By comparison, there are only about 1080 atoms in the universe. If OpenVaccine players considered one candidate every second, it would take longer than the life of the universe to get through them all.
Every time an OpenVaccine player changed a codon on an RNA vaccine they were building, LinearFold would compute both the structure of that sequence and how much "free energy" it had, which is a measure of stability (lower energy means more stable). The runtime for each computation was about 3 or 4 seconds. The players came up with a number of interesting candidates, a few dozen of which were synthesized in labs for testing. But it was clear they were exploring only a tiny number of the possible candidates.
The LinearDesign algorithm, which my group completed and released in April 2020, comes up with RNA sequences that are optimized for stability and that rely on the body's most used codons, which leads to more efficient protein production. (We published an update with experimental data just this week.) As with LinearFold, we made the LinearDesign tool publicly available. Today, OpenVaccine players by default use LinearDesign as a starting point for their exploration of vaccine candidates, giving them a jumpstart in their search for the most stable sequences. They can quickly create stable structures with LinearDesign, and then try out subtle changes.
 This “wildtype" RNA structure (that found in the natural coronavirus) codes for the production of the spike protein, but it contains a number of loops with unpaired nucleotides, making the structure less stable. Our LinearDesign algorithm produced many structures with far fewer loops; importantly, the RNA still codes for the spike protein. Huang Liang
This “wildtype" RNA structure (that found in the natural coronavirus) codes for the production of the spike protein, but it contains a number of loops with unpaired nucleotides, making the structure less stable. Our LinearDesign algorithm produced many structures with far fewer loops; importantly, the RNA still codes for the spike protein. Huang Liang
My team has also used LinearDesign to produce vaccine candidates, and we're working with six pharmaceutical companies in the United States, Europe, and China that are developing COVID-19 vaccines. We sent one of those companies, StemiRNA of Shanghai, seven of our most promising candidates for COVID-19 last year. Those vaccine candidates are not only confirmed to be more stable, but also have already been tested in mice, with the exciting result of substantially higher immune responses than from the standard benchmark. This means that with the same dosage, our vaccines provide much better protection against the virus, and to achieve the same protection level, the mice required a much smaller dose, which caused fewer side effects. Our algorithm can also be used to design better RNA vaccines for other types of infectious diseases, and it could even be used to develop cancer vaccines and gene therapies.
I wish that this work on analyzing and designing RNA sequences had never become so crucial to the world. But given how widespread and deadly the SARS-CoV-2 virus is, I'm grateful to be contributing tools and ideas that can help us understand the virus—and overcome it.
From Your Site Articles
- This Is How We'll Vaccinate the World Against COVID-19 - IEEE ... ›
- Pfizer's Edge in the COVID-19 Vaccine Race: Data Science - IEEE ... ›
- Inovio's Electrical Device Zaps a COVID-19 Vaccine Into the Body ... ›
- The Algorithm that Mapped Omicron Shows a Path Forward - IEEE Spectrum ›
Related Articles Around the Web

