
The international Human Genome Project and the private genomics company, Celera Genomics, of Rockland, Md., plan to publish the first draft of the entire human gene sequence early next year. This has been a top flight engineering achievement. A single DNA sequencing machine today can produce over 330 000 bases (units of sequence information) per day, more than 100 researchers could manage in a year and a half using manual techniques less than a decade ago.
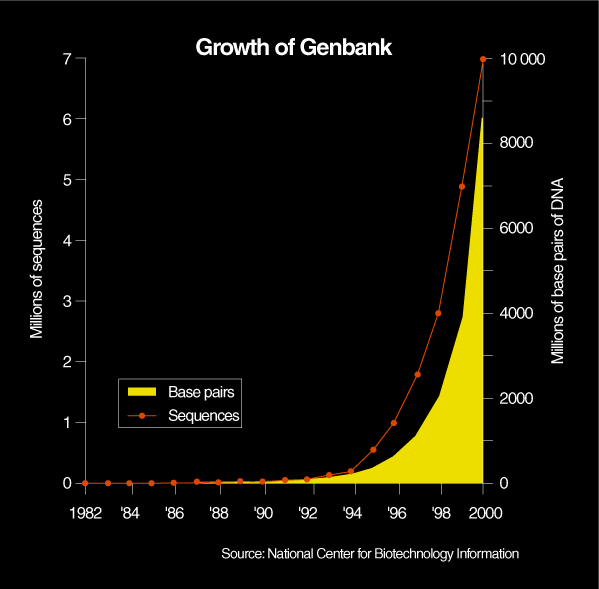
Major genome centers such as the Sanger Center in the UK or the Whitehead Institute for Biomedical Research, in Cambridge, Mass., run over 100 such machines. Celera, with 300 machines, has a production capacity approaching two billion bases per month, or more bases than researchers had identified in the 16 years between 1982, when central databases began, and 1998 [Fig. 1]. Moreover, the reliability and speed of the automated methods that became available in the past two years have increased to such an extent that high-powered computational approaches can be applied to the reconstitution of the human genome sequence from millions of fragments of processed DNA.
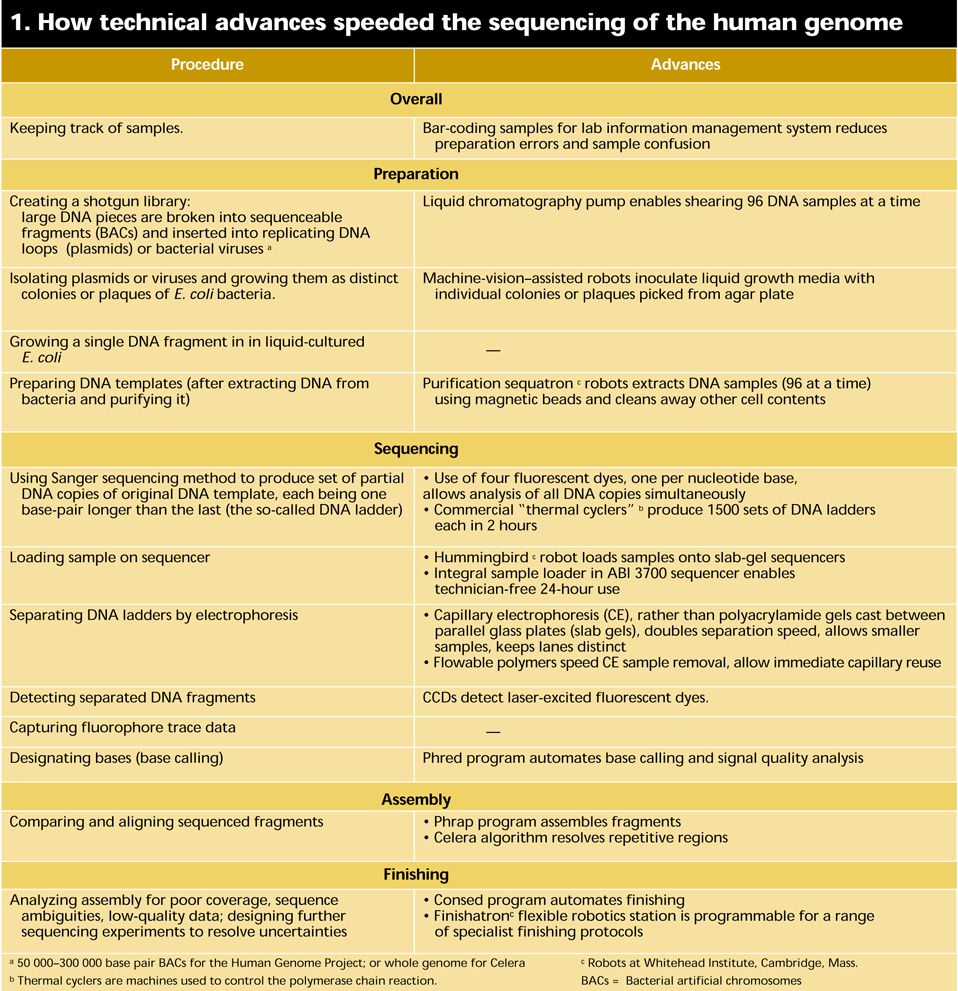
At a basic level, DNA sequencing calls for the preparation and replication of relatively short segments of DNA; the creation of partial copies of the segments each one base longer than the next; the identification of the last base in each copy; and the ordering of the identified bases. At each step technological developments have accelerated the pace of discovery [Table 1]. Industrial robots shift genetic material from station to station with speed and accuracy. Sequencing machines using multiple capillaries filled with polymer gels have catapulted throughput to new heights and reduced a major source of sequencing error. And DNA assembly computer systems have been constructed that recently set in order a 120-million-base genome in less than a week’s worth of calculations. For more background on the status and meaning of the Human Genome Project, as well as the definitions of terms in this article, see “Understanding the human genome.”
A slow start
The history of the sequencing of the set of human genes has been one of plans being overtaken by technical events. That, in many ways, was inevitable and in fact it was expected. The original vision for the U.S.-funded Human Genome Project was a 15-year program begun in 1990, the first 10 years of which would be deployed in constructing comprehensive maps of the genome and in developing a new generation of sequencing technologies. In 1990, all DNA sequencing was performed using the method developed by Frederick Sanger at the Laboratory of Molecular Biology in Cambridge, UK, in 1977.
At the fundamental level, nothing much has changed 10 years on: Sanger sequencing is still the method of choice. But how the technique is practiced is almost unrecognizable.
Sequencing in 1990 was expensive, at around US $2-$5 per finished base, the basic unit of genetic information, and indicated a cost for the whole project of $6 billion to $15 billion. The figure needed to come down to around 50 cents a base to meet government funding requirements. Leroy Hood, co-developer of the first generation of sequencing robots while he was at the California Institute of Technology, said in 1993 that he would be “shocked” if sequencing costs had not fallen to 10 cents per base by the time the human genome was completed. He was right.
At that time a number of techniques were being developed with an eye to breaking through to the 50-cent-per-base goal. They included capillary gel electrophoresis, the incorporation of stable isotopes of nitrogen or carbon into DNA and their detection by mass spectrometry, X-ray imaging, and physical techniques such as scanning tunneling or atomic force microscopy. In the end, however, only capillary gel electrophoresis, a technological tweaking of Sanger’s original recipe, came through in time.
For a while, genome researchers were content to use nonautomated methods for gene sequencing. To produce the bulk of the first complete eukaryote chromosome, baker’s yeast chromosome III, researchers cast electrophoresis gels by hand, loaded them by hand, and eye-balled the result. By the time the complete, 16-chromosome yeast sequence was published in 1996, manual sequencing was almost extinct.
The yeast work had involved over 100 laboratories, in part because political pressures within Europe resisted attempts to focus large amounts of public research money in just a few centers. The U.S. Human Genome Project was also prey to such sensitivities. The project’s early budget was directed at establishing 10 or 20 sequencing centers around the United States that would be part of a consortium approach. Some saw that as a political solution to counter potential complaints over the distribution of funds. In any event, four U.S. centers—Washington University in St Louis; the Whitehead Institute in Cambridge, Mass.; Baylor College of Medicine in Houston; and the Joint Genome Institute in Walnut Creek, Calif.— together with Sanger Center near Cambridge, UK, have generated the bulk of the sequence data in the publicly funded project. At least part of the impetus for this concentration of effort at a few high-volume specialist centers came as a result of the work of Daniel Cohen, former scientific director at a laboratory called Généthon, in Paris.
Beginnings of automation
Cohen’s was the first molecular biology laboratory to throw automation at a problem in a significant way. It could do so simply because of its unique funding mechanism. A charity, the French Muscular Dystrophy Association, spent $7.5 million to found what was then the world’s largest genomics laboratory, Généthon, in 1989. Généthon’s aim was not to sequence the human genome but to produce a physical map—an ordered set of large blocks of unsequenced human DNA. A number of laboratories had produced human DNA libraries, consisting of thousands of DNA segments held as separate genetic elements in organisms like yeast. But such libraries were like a collection of separate pages ripped at random from several copies of the human book of life. Cohen planned to put those pages back in order.
At a time when few labs had more than one DNA sequencer, Généthon had 18 machines, each outputting an average of over 8000 bases per day. The laboratory also ran thousands of samples through the polymerase chain reaction (PCR), a technique used to amplify a specific region of DNA. PCR in turn requires thermal cyclers, which are Peltier-based reaction heaters that switch the samples dozens of times between two temperatures—the one at which DNA is replicated and a second at which its two strands peel apart. Généthon had more than 20 standard PCR machines and three prototype devices, each handling 1500 DNA samples at a time.
At the heart of Généthon’s effort, however, were two custom robots designed and built in-house. Saturnin was a spotting robot that could place 450 000 samples of DNA per day on a membrane for reactions. Barbara was a clone-sorting robot used, amongst other things, to sort Généthon’s clone library for distribution to the scientific community. Key to virtually all automation systems in the human genome project is the microtiter plate, a rigid polycarbonate receptacle with 96 (or, latterly, 384) equally-spaced wells, U-shaped in cross section. The use of such plates allows movement of multiple microliter samples with sure knowledge of their location. Cohen’s Saturnin robot could be loaded with over 600 microtiter plates containing 55 000 addressable sample positions in all.
When Généthon published its physical map of the human genome in 1993, it generated a reference resource that other researchers could call on to organize their work. More than that, though, the way in which it had generated the map altered the technical focus in other parts of the genome research community. According to Cohen, Généthon had considerable influence on the development of both the Sanger Center, which was just being built in 1993, and the Whitehead Institute’s Center for Genome Research. At the end of 1995, Whitehead and Généthon produced another, finer physical map wherein—to continue the “book of man” metaphor—the pages had been subdivided into paragraphs and reordered. Significantly, of a two-and-a-half year project, the first 18 months had been devoted to the development of automatons for sample handling and of an information management system that allowed the tracking of samples around the whole system.
By 1998, the goals of the U.S. Human Genome Project had changed somewhat, most strikingly in target date. Completion of the sequence was now scheduled for 2003, two years earlier than had been expected—and 50 years after James Watson and Francis Crick elucidated DNA’s structure. The project had clearly become an international one. Predictions were that U.S.-funded activity would now contribute only 60-70 percent of the results and the remainder would come from the Sanger Center, funded by the Wellcome Trust in the UK and other centers. Completion by 2003 was seen as a highly ambitious, even audacious goal, given that by 1998 only around 6 percent of the human sequence had been completed.
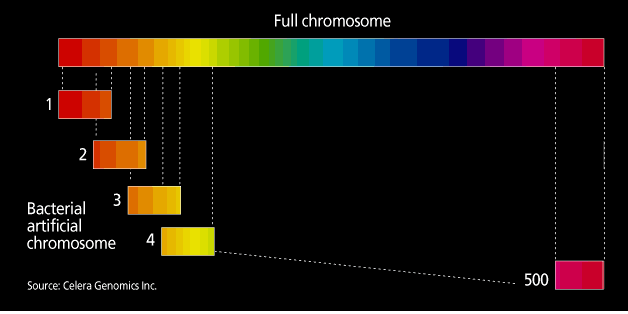
The project and the private company Celera are taking different tracks to sequencing the genome. In the publicly funded genome projects, the approach is a systematic generation of sequencing information, termed clone-by-clone. In essence, mapping divides the human genome into manageable fragments whose order along the genome is known. The various centers are now working with bacterial artificial chromosomes (BACs), fragments containing human DNA inserts of 50 000-300 000 bases. At each sequencing center, mapping groups identify a so-called tiling path of minimally overlapping BACs, to enable sequencing of the genome to occur in as close to a single pass as possible [Fig. 2]. Each BAC in a given region of the genome is then sequenced using what had been dubbed a shotgun approach, another of Fred Sanger’s innovations.
In principle, shotgun sequencing is simple. Each BAC is broken down further into thousands of overlapping fragments — the BAC library— the sequence of each of which can then be determined in a sequencing machine. The sequence regions common to overlapping fragments are used to align them and reassemble the full sequence of the original BAC. But in practice, the process is complicated by the sheer number of samples involved. An average BAC will be reconstructed from 2000 to 3000 separate fragments from the library.
“Library making is probably the least automated of the processes that we do,” said Elaine Mardis, who heads up the technology development group at Washington University in St Louis. “There is no library-making robot.” The only step that has been automated anywhere in the process is DNA fragmentation, where specially adapted pumps are used to shear DNA samples contained in a 96-well plate. DNA pieces of 1000 to 2000 bases are then inserted into carrier molecules, such as loops of bacterial DNA (plasmids) or the DNA of the M13 virus for replication. The samples are then ready for the production phase, which is where most efforts at streamlining and automation have been focused. Production is “like bottling beer or making cars,” said Mardis. “It’s a very reproducible task.”
The first stage is picking the subclones—bacteria used to multiply the BAC library. Robots with machine vision systems access bacterial colonies growing on an agar plate and place them into liquid growth media in 96-well microtiter plates. Each robot does this 2000 times per hour. At the Sanger Center, the job is perfomed by a team of five colony-picking robots built by the center’s own engineering department.
The DNA from each clone then has to be isolated and purified, a transitional task between the biology of clone production and the chemistry of sequencing. It is essentially a two-step process: the separation and cleaning of the DNA from the other cell contents, followed by the amplification of the inserted DNA fragment.
Separating and cleaning DNA is a slow process whose rate is dependent on getting DNA efficiently out of solution following a series of washes. Elaine Mardis’ group is in the process of upgrading the Washington University DNA separation capability by copying a design developed at Eric Lander’s laboratory at the Whitehead Institute [Fig. 3]. That technology accelerates the precipitation of DNA by attaching the molecules reversibly to microscopic magnetic beads. The Whitehead robot is, in essence, a 7.5-meter conveyor belt that takes the samples past a series of stations for either reagent addition or magnetic separation. At each magnetic station, the beads (and hence the attached DNA) are drawn into a circle so that the surrounding liquid impurities can be sucked out by a pipette tip placed in the center.

Sequencing strides
Inserted DNA is simultaneously amplified and sequenced using the polymerase chain reaction, a replication method that won a Nobel Prize but is now a routine laboratory procedure. For each sample, a set of DNA fragments is produced, each one varying in length by a single base and each labeled with one of four fluorescent dyes, which correspond to the the last base in the fragment. At Sanger, that amplification is performed on 1536 samples about every 2 hours. The amplified sequences need to be cleaned again to remove PCR reagents before being stored at -20 °C to await the sequencing phase itself.
“That’s when the samples come to me,” said Chris Clee, group leader of the sequencing team at the Sanger Center. Clee has some 200 sequencing machines occupying most of the bench space in three adjoining laboratories. Sixty of them are so-called slab-gel sequenators, 377s from Applied Biosystems Inc. (ABI), Foster City, Calif., a subsidiary of PE Corp. The other machines are more advanced capillary electrophoresis-based, either ABI 3700s or MegaBaces from Molecular Dynamics Inc. of Sunnyvale, Calif. (a division of Amersham-Pharmacia Biotech Ltd., Uppsala, Sweden).
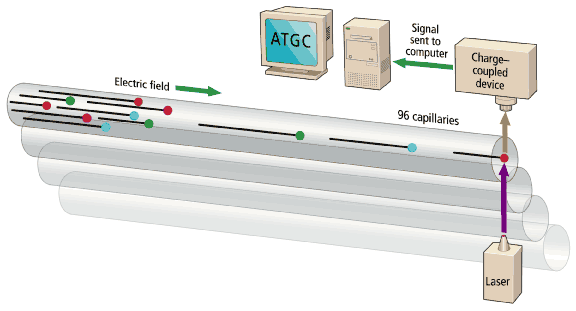
The sequencing machines are, in essence, multitrack electrophoretic separators [Fig. 4]. DNA fragments in the sample bear a uniform negative charge per unit length, but as they migrate in an electric field, the gel matrix slows large fragments to a greater extent than smaller ones. The time of migration thus indicates the size of the fragment. The reading of DNA fragments from a few bases up to 500-600 bases long will take 5-6 hours in slab gel or 2-3 hours in a 50-cm capillary. The 500-600 base fragments are near the maximum length of analyses in high-throughput production sequencing. Reads beyond 1000 bases are possible, but take longer, and signal quality is lost.
The ABI 377s were the main workhorses of the genome projects until early 1998. They are slab-gel machines onto which 96 samples are loaded, usually by manual pipetting into 96 evenly spaced notches at the top of the gel. The DNA samples migrate in parallel tracks down the gel and are detected as they emerge by a fluorescence detector. Their big disadvantage was the fact that they used slabs of gel, which have to be prepared manually close to the time they are needed.
“Gel quality varied from batch to batch and depended on who was preparing it,” said Chris Clee. “Even the time of year made a difference. Gels were better during the summer because of the lab temperature, although sometimes that affected the water quality.”
Quality control was not the only problem. Running 96 lanes close together often confused the optical systems of the sequencing machines, with the result that “reads” would be taken from adjacent lanes. This could throw the whole sample-tracking system out. Members of the sequencing “finishing” team, whose job was to compare overlapping regions from the analysis, would have to compensate for this by looking at reads on either side of the lane they thought they should be examining.
Both those drawbacks disappeared with the emergence of the capillary electrophoresis-based sequencers. Although capillary electrophoresis (CE) had been discussed as a sequencing option before the start of the Human Genome Project in 1990, the first CE-based machine, the ABI 310, was launched only in 1995. The 310 contained a single capillary and was suitable for low-volume research applications, rather than production of vast amounts of DNA sequence. Amersham-Pharmacia Biotech beat ABI to the industrial-scale punch with the launch in 1997 of the MegaBace, a 96-capillary machine onto which the contents of a whole microtiter plate could be loaded. The MegaBace has been significantly outsold by the ABI 3700 at the major genome production centers, partly because ABI is the market leader and partly because its machine has an integral sample-loading robot. This can hold two 384-well plates, enabling a day’s worth of sequencing runs to be loaded at once. “As long as things go smoothly, you don’t really have to attend to it until the next morning,” said Elaine Mardis. “With the MegaBace, you have to go back to it every 3 hours and reload. The machine attendance time is only 15 minutes, but you still need someone there to do it.”
“The 3700 was designed as walkaway automation,” said Michael Phillips, who was the engineering program coordinator at ABI for the development of the 3700. He points to two key developments that made that possible: one is automatic loading and the other is the development of suitable flowable polymer matrices.
After the operator inserts two 384-well microtiter plates, the machine does the rest. A robotic arm takes samples two at a time and places them on a loading bar. At that point, the sample sits just at the end of the capillary. When 96 samples have been loaded, the device applies a low voltage to draw the sample down into the capillary. The high-voltage field is then turned on. In due course, the samples emerge at the bottom of the 50-cm capillaries where they flow into a stream of polymer. A laser directed into the polymer stream enables the detection of the 96 separate outputs.
The polymer matrices were probably the real key to walk-away automation for the 3700. What ABI wanted was a polymer that could be flushed away after each run. The crosslinked polyacrylamides used in the slab-gel approach were never going to be suitable for CE: they adhere too well to the capillary walls. “Samples would have to be run all the way through the gel before reloading could occur,” said Phillips. “That would have cost a lot of time in each run and it would have meant that the capillaries would have to be changed after four or five runs.” On the other hand, the polymers available in the early ’90s tended to be drawn down the capillary by the extremely high voltages used in capillary CE, interfering with the resolution of the DNA fragments. The proprietary polymers that ABI developed in the mid-’90s do not migrate in the strong electric fields but can be readily pumped out after electrophoresis.
The capillary configuration means that the lane confusion seen in slab-gel sequencers is eliminated. Furthermore, the flowable gels used in capillary electrophoresis are quality controlled by the manufacturer. The improvement in performance is not just counted in data throughput (although there are marked improvements there) but also in data quality.
When it works well, the automatic loading system is a significant benefit for “power sequencing” laboratories. But robotic failures are one of the most common causes of sequencer down-time for the 3700. Chris Clee at the Sanger Center estimates that he might lose 5 percent of capacity on an average day because the ABI loading system is not working properly. ABI’s Phillips suggests that that particular issue may recede, now that ABI has pinned down the main cause to a factor outside the sequencer itself. “The microtiter plates were warping,” he said. “For certain brands of plate, the repeated exposure to hot temperatures during thermal cycling put plates slightly on the skew—nothing an operator would notice, but the machine with its lower tolerances did.” When the company diagnosed the problem sometime later, the engineering staff fixed it simply with a clamp to hold the plate down.
Assembly Algorithms
The computational process for assembling each shotgunned BAC is relatively straightforward and starts once 500 of the 2000 to 3000 reads performed for each BAC have been completed. It is repeated every time another 200 reads are added out of the sequencer. The standard genomic assembly software is a suite of three programs, Phred, Phrap, and Consed, developed by Phil Green, a professor of molecular biotechnology, and colleagues at the University of Washington Genome Center in Seattle.
Phred (Phil’s read editor) looks at the logged and stored fluorescent signal output from the DNA sequencers, identifies (or calls) the bases, and assesses the quality of the signal for each base. Phred assumes that the emergence of the DNA fragments will create an ideal set of sharp, evenly spaced fluorescence signals. Using simple Fourier methods, the program then assesses, for each detected base, how far reality departs from this ideal. The shift in the real signal from those ideal positions due to uneven migration, local loss of signal intensity, or other factors is returned as a logarithmic quality score. Thus, for data of Phred 15 quality, there is only a one in 1015 chance that the base call is wrong.
Phred’s companion program, Phrap (for phragment assembly program, or Phil’s revised assembly program) assembles the shotgun DNA sequence data by overlaying different reads and looking for identity among them. It uses Phred scores to guide it to the highest-quality data, and lets the human operator know about its decision-making processes to aid trouble-shooting. Through this overlaying process, assemblers gradually build up longer and longer gap-free stretches of data.
Phrap interacts very closely with Consed, a consensus visualization and editing program that can be used to guide a human finisher. Finishers examine the quality of sequences from overlapping regions and determine what new sequencing runs are required to improve data quality in particular regions or to fill sequence gaps. Through a set of visual highlighting mechanisms, Consed helps the finisher find problem areas in the assemblies made by Phrap: these might be areas of sequence mismatch, or genome regions where little sequence information has been gathered because only a single clone has been sequenced there. The finisher can scroll through the sequences quickly, identify which reads or which clones are responsible for the poor data, and annotate the data appropriately. Consed will also suggest what kind of further work, such as resequencing, might have to be done. The Autofinish module of Consed can reduce the decision burden for the human finisher by 85 percent.
For the public sector genome projects, the computational burden is not large. Of course, the right hardware, interfaces, and software updates have to be in place. Where it certainly is critical, however, is in the whole-genome shotgun approach that Celera Genomics is undertaking. Celera has shunned the idea of pre-dividing the genome into bite-sized pieces and methodically sequencing one after another. Instead, it is producing sequence reads from a whole genome library of 40 million DNA fragments and then assembling those at one go. According to Eugene Myers, vice president of informatics research at Celera, assembly programs like Phrap can cope with problems that involve the assembly of 2000-40 000 fragments but not with 40 million reads.
Whole genome shotgun sequencing was first performed on the bacterium Haemophilus influenzae. One of the biggest difficulties in going from bacterial sequence data to such eukaryotes as the fruit fly and humans is not simply the sheer size of the genome, but the inconvenient presence of repetitive elements of DNA distributed throughout it—inconvenient in that the elements are far longer than the maximum read length from a sequencer. At the accuracy expected from the sequencing reactions, repetitive elements from one part of the genome are essentially indistinguishable from those in distant parts, creating a huge risk of misassembly.
Celera addressed the repetitive DNA problem by producing mates, DNA fragments long enough to span the repetitive elements. The fruit fly, Drosophila, served as the company’s 120-million-base sequencing guinea pig. For it, Celera produced three sets of mates: 654 000 with spans of around 2000 bases, 497 000 with 100 000-base spans, and over 12 000 BAC mates where the separation of the end was around 130 000 bases. Both ends of the mates were sequenced for 550 bases or so and, together with sequence data from nonrepetitive regions, the data was turned over to Celera’s proprietary assembly program.
The assembly algorithm at Celera takes advantage of what the system knows about the shotgun process. It tentatively puts regions of DNA together based on raw sequence data comparisons and then assesses whether that assembly is likely. “For example,” explained Myers, “if we’ve done 10 times data collection, then the assembled fragments should be stacked up about 10 deep. If they’re stacked up 100 deep, then it’s probably not correct.” The statistical cutoff applied to the data meant that the error rate of such an assembly method would be only one in 1015. “We would then take those subassemblies that we were 100 percent certain of and use mate pair information to link those together. What we were able to find is that you get almost the whole genome doing that.”
The computing power that Celera needed to store the vast amount of sequence information and to analyze it is nearly an order of magnitude greater than that at any of the publicly funded centers. The Sanger Center, for instance, has around 250 Compaq Alpha Systems backed by over 4.5 terabytes of disk storage. Celera’s supercomputing facility, in contrast, currently houses 848 interconnected Alpha processors and has 50 terabytes of disk storage capacity.
Even computing power at that level was challenged by the task of assembling the human genome from 40 million fragments. Celera had assembled the fruit fly genome from over three million DNA fragments in one batch process taking less than a week’s worth of computer time. The human genome is 30 times bigger than the fly’s: Myers calculated that it would have required a prohibitive 600GB of memory to undertake the human genome assembly in the same way.
“I don’t know of any machines that give you a single memory of that size,” he said. “So we had to figure out a way to get around that.”
One thing Celera did was to reduce the number of DNA fragments it processed. For the fruit fly, the company assembled an amount of DNA equivalent to 13 times the entire fly genome. Subsequently, it found it could get almost as good a result with half that coverage. “For the human genome, we realized we’d have a good assembly at 6.5 times. So we changed strategy a little bit.” explained Myers.
Celera also departed from the single batch processing of the fly genome assembly. Instead, the assembly program was fed partial data from which it constructed little subassemblies that it was sure about. Then it cleared out the unneeded data from fully established overlaps, kept the subassemblies, and sought more fresh data. “The human assembler is continually updating its internal state in response to the arrival of data,” said Myers. “In this way, we reduce the size of the computational problem by about a factor of 30. We were able to do the human genome in 32GB of memory, which is pretty phenomenal when you consider that it took 20GB to do Drosophila.”
Next Developments
The completion of the human genome project does not represent an end for genomics. The human sequence still contains gaps and probably will not be complete for at least another two to three years. Furthermore, there are lots of genomes of other important species to be sequenced. Celera recently finished sequencing the mouse genome, and other groups will eventually turn to farm animals and possibly crop plants. The various large genome centers, no longer bonded by a common grant or cause, are likely to disperse intellectually and pursue separate lines of activity. The Whitehead Institute and the Sanger Center plan to build on their automation capability, and become resource centers for DNA-microarray analysis, performing highly multiplexed routine genetic analysis as part of large studies to track the effects of genetic variation within human populations. Celera has ambitions to become a commercial genetic hub in the e-health revolution.
The sequencing machines will evolve with them. ABI is preparing to launch a series of capillary electrophoresis machines designed not for production but for rapid analyses taking less than 40 minutes. Sequencing will become resequencing or comparative sequencing—the search for genetic differences between individuals.
To Probe Further
For a more technically detailed view of automation in genomics, see Deirdre Meldrum’s “Automation for genomics,” Parts 1 and 2, in Genome Research, Vol. 10, pp. 1081-92 and pp. 1288-303, respectively.
“A Whole-Genome Assembly of Drosophila,” by Eugene Myers and others in Science, 24 March 2000, pp. 2196-204 gives a thorough description of Celera’s whole genome assembly process.